We use symmetry-adapted perturbation theory based on density functional theory (SAPT-DFT) to derive accurate interaction potentials of molecular hydrogen with carbon nanostructures (graphene/graphite and single-wall carbon nanotubes, SWCNTs) [1-3]. Small hydrogen-saturated carbon clusters are used to obtain and fit dispersion- and dispersionless-interaction parameters. These parameters are then used in a pairwise model to obtain the full interaction of H2 with extended nanostructures. The resulting potentials are also used to assess the accuracy of different DFT van der Waals-corrected functionals [1].
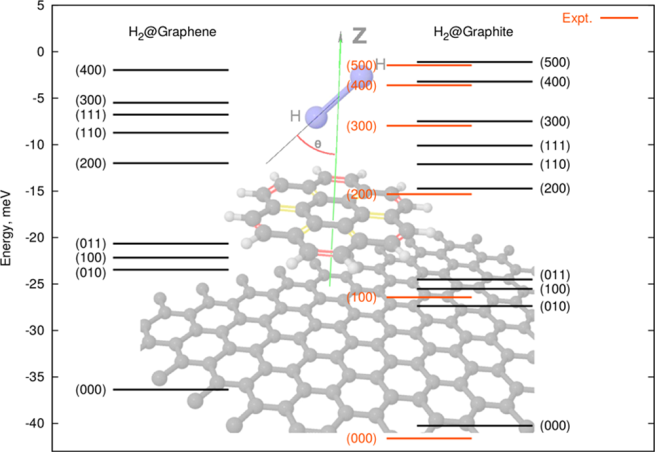
The interaction potentials are further applied to study the quantum motion of up to four H2 molecules confined to different nanostructures [2]. For a single molecule, the molecular rotation is explicitly taken into account. The calculated vibrational levels of H2 on graphite [1] (which is a simple assembly of graphene sheets) are in extremely good agreement with the experiment, see Fig.1. The rotational splitting (m=0,1) of (j=1) ortho-hydrogen [3] is of the correct order of magnitude, but somewhat larger than available experimental data obtained for an assembly of SWCNTs. This difference might be due to the orientational damping and requires further investigation.
For clusters of hydrogen molecules [2], the structure-less (j=0) boson model is used, that is justified due to the higher rotation energy (2B=120 meV) of H2 as compared to the H2-nanostructure interaction (about 10 meV). We have shown that three H2 molecules form a triangular structure inside (11,4) SWCNT subject to a very large amplitude motion, see Fig.2. To obtain the corresponding wave functions and energies, we used the discrete-variable-representation (DVR) numerical approach in internal coordinates. The obtained arrangements are in line with very recent experimental data, indicating the stability of HCP structure of hydrogen stored inside SWCNTs and pointing out their very strong quantum nature. Work is in progress to treat larger molecular clusters at ab initio quantum level.
References :
1. M.P. de Lara-Castells, A. Mitrushchenkov, J.Phys.Chem.A 119, 11022(2015)
2. M.P. de Lara-Castells, A. W. Hauser, A. Mitrushchenkov, and R. Fernandez-Perea, Phys. Chem. Chem. Phys. 19, 28621 (2017)
3. M.P. de Lara-Castells, A. Mitrushchenkov, Phys. Chem. Chem. Phys. 21, 3423(2019)
| Subject : | : | poster |
| Topics | : | Quantum N-body problem in theoretical chemistry |
| Topics | : | Poster session |
| Keywords | : | hydrogen molecules ; carbon nanotubes ; graphene |
| PDF version | : |  PDF version PDF version |
- Picture